Leader investigator: Joanna Szyda
Period of work: 36 months
Funded: National Science Centre
Objectives
The aim of the project was to a better understanding of genetics underlying hoof diseases and lameness. Traits related to hoof diseases and lameness are called novel traits since their phenotypes have been collected only recently, and thus their genetic background has not yet been extensively studied. However, this group of traits play important role in breeding and management. Lameness can cause economic losses due to lower production and fertility. Therefore, the objective of the project is to analyze the genetic determinants of traits describing hoof and leg quality in the context of the causes lameness of dairy cattle, with particular emphasis on epistatic effects. Traits inclueded in this project were : hoof health status defined by a veterinarian (HSV) (heritabilitiy of 25%), by a claw trimmer (HSC) (heritabilitiy of 28%), and the total number of hoof disorders (NHD) (heritabilitiy of 8%).
The dataset consists of hoof and leg quality trait values and 76 934 SNP genotypes for 985 Braunvieh cows and 1998 Fleckvieh cows. Moreover, we also have the information about SNP locations, pedigree and culling of individuals (the number of culled animals ad culling reasons), some additional variables such as the date of birth and the number of finished lactations are available as well. The data set was made available by the courtesy of ZuchtData EDV-Dienstleistungen GmbH.
The preliminary analysis of available data showed that 2.04% all culled cows and 4.80% of cows culled due to health problems or low productivity were removed from herds because of hoof hood or leg disorders. The main causes of culling are age (48.97%), low fertility (24.50%) and udder diseases (13.07%).

fig1. Significant epistatic effects for the number of claw disorders.

fig 2 . A,B before removing polymorphisms with the call rate below 95% and polymorphisms with the minor allele frequency below 5%. C,D after removing polymorphisms with the call rate below 95% and polymorphisms with the minor allele frequency below 5%
The analyzed data set contain iformation of hoof disorders scored for Braunvieh and Fleckvieh cows durring different parities within the frame of the “Efficient Cow” project. For each individual, three lameness-related traits collected scored until 100th of lactation: hoof health status defined by a veterinarian (HSV) [binary], hoof health status defined by a claw trimmer (HSC) [binary], and the total number of hoof disorders (NHD) [discrete values between 0 and 5]. 2,977 cows (including both breeds) had records for HSV trait and 1,513 for HSC and NHD. For each individual breeding values were estimated (EV). Each individual was genotyped with the GeneSeek® Genomic ProfilerTM HD panel consisting of 76,934 SNPs. After quality contror of a minor allele frequency (MAF) of at least 0.01 and technical quality of genotyping expressed by a minimum call rate of 99%, 74,762 SNPs remained for further analysis.

fig.3 The indicidents rate of found nr of hoof disorders in two cattle populations from none leg disdorder:0, to more then five: >=5
We applied first-stage genome-wide analysis study (GWAS) usuing SNPs from HD panel. Second-stage GWAS was done based on SNPs from whole genome sequence, where significant region identied from first-stage GWAS were imputed separately for each breed using Beagle 4 software. We also performed a GWAS were phenotypic values were represented by pseudophenotypes, which were estimated breeding values (EV) for each individual estimated for the total number of leg disorders. EV were estimated by using ASReml software.
Based on the first-stage GWAS with SNPS from the HD panel, seven significant genomic regions were defined identified: BTA1 including the TOPBP1 gene; BTA7 including the RIOK2 and RGMB genes; BTA13 including the C13H20orf194; regions on BTA14 which inclued RRM2B and NCALD genes on first and STK3 gene on second region; BTA15 including FAM168A and PLEKHB1 genes; BTA22 including PTPRG gene. Based on the results from second-stage GWAS 23 SNPs were defined as significant located region on BTA15. Results from last GWAS where as phenotypes were resperented by pseudophenotypes of EV, showed that Based on the false discovery rate of maximum 10%, 16 SNPs located on BTA1, BTA4, BTA5, BTA6, BTA13, BTA14, BTA16 and BTA24.

fig.4 The X-axis marks SNP positions in bp, the Y-axis represents –log10(P), visualize genomic context was done for the most significant SNPs for hoof HSC mapped to ARS-UCD1.2 assembly.
Genomic regions with significant SNP effects on hoof health status and potential candidate genes were found, throughout different analysis.
NCN grant No. 2015/19/B/NZ9/03725 as well as by the Efficient Cow and the Gene2Farm projects
“Functional Annotation of Animal Genomes – European network (FAANG-Europe)”
Period of work: May 2016 – 30 April 2017
Funded: COST Action
Involved countires: Bulgaria, Croatia, Denmark, Germany, Greece, Ireland, Malta, Netherlands, Poland, Portugal, Serbia, Slovenia, Spain, United Kingdom and Macedonia.
The main goal of this grant is improving the functional annotation of animal genomes in order improve the accuracy of predicting phenotypes from genotypes (sequence). Understanding the regulation of gene expression in the target species will facilitate the development of predictive models based on understanding of the underlying biological mechanisms. Identifying regulatory sequences and the consequences of variation in such sequences are important for the development of the improved predictive models.
In details, areas of expertise relevant for the action are :
Key investigators: Magda Mielczarek, Joanna Szyda
Period of work: 2015-2016
Funded: National Science Centre
Objectives: Copy number variations (CNVs) are the major source of genetic diversity in mammals and they are defined as the gains (duplications) and losses (deletions) of the DNA fragments. Their length ranges from 50 bp to several milions bp and they are present widely in genomes. Moreover, CNVs cover many functional elements of the genome, such as genes or regulatory sequences which can markedly effect the phenotypic characteristics of individuals. In the project we have a unique opportunity to analyse CNVs across many individuals belonging to 9 different breeds of domestic cattle (Bos taurus Linnaeus, 1758). Therefore, the main aims of this study comprise (i) detecting CNV polymorphisms in 121 individuals, (ii) describing the distribution of these polymorphisms across the genome (iii) assessing the inter-breed and inter-individual variation in the number and distribution of copies, (iv) the functional annotation of CNVs as well.
Methods: The way leading to obtain a reliable set of annotated CNVs included the following steps: (i) an alignment to the reference genome, (ii) data processing after alignment, (iii) CNVs detection, (iv) validation of CNVs and (v) their annotation. The final set of validated CNVs was subjected to the statistical analysis in order to provide population wide inferences. In particular, separately for duplications and deletions the inter-individual and the inter-breed variation analysis of the number and the distribution of detected polymorphisms were tested.
Results:
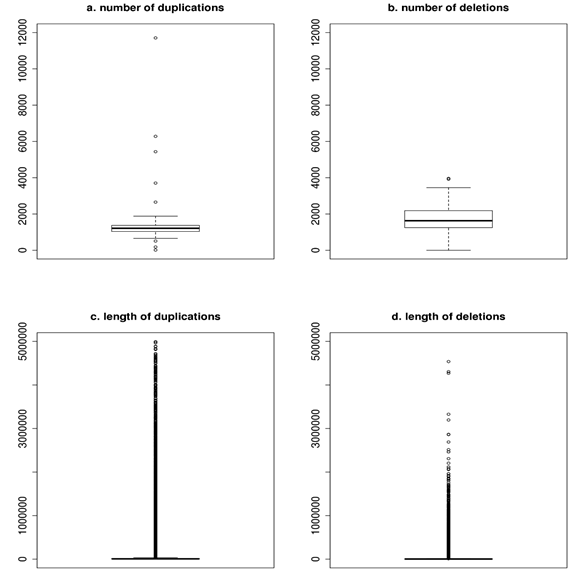
In order to create the final and realiable dataset of polymorphisms overlapping CNVs were determined. The number of duplications per animal ranged between 12 and 11,704, while mean and median were 1,343 ± 1,086 and 1,212. The number of deletions ranged between 0 and 3,960 and the corresponding mean and median were 1,708 ± 700 and 1,628. The length varied strongly and for duplications ranged from 200 to 4,992,800 bp. The corresponding mean and median were 31,018 ± 169,307 and 6,900 bp. The shortest deletion was 200 bp and the longest 4,536,800 bp. The mean was 10,836 ± 53,724, while the median was equal to 2,000 bp. The graphical representation of the number of duplications (a) and deletions (b) per bull and the length of duplications (c) and deletions (d) observed in the whole validated data set is presented in figure below.
As the effect of the functional annotation 70.51 % of duplications and 67.92 % of deletions fell into non-genic regions while for 29.49 % of duplications and 32.08 % of deletions SO terms corresponding to gene regions were assigned. The figure below shows the proportion of both annotation types across all duplications and deletions.
6. Conclusions
In the present study, the genome assessment of Copy Number Variations in cattle using whole genome sequence data was performed. The analysis enabled detection, validation and annotation of 445,791 CNVs as well as hypotheses testing of obtained results. A highly significant inter-individual and inter-breed variation was observed both in the number and in the length of CNVs. The breed-specific phenomenon was especially strongly emphasized for the Fleckvieh breed suggesting it was subjected to different artificial selection pressure than other breeds. Moreover a significant variation in the CNV distribution was also observed within a genome by varying density of CNV depending on genome function. Summarising, the analysis showed a high complexity of a CNV landscape in Bos taurus genomes.
—————————————————————————————————————————-
Results included here were essential to write the PhD thesis “The genome-wide distribution of copy number variations in various breeds of domestic cattle (Bos taurus Linnaeus, 1758) based on the next-generation sequencing data”. The PhD thesis was defended on 10.10. 2016, on the Faculty of Biology and Environmental Protection at Nicholas Copernicus University in Toruń.
Pełny tytuł: Next generation European system for cattle improvement and management
Czas realizacji: 48 miesięcy
Numer wniosku: 289592-Gene2Farm
Instytucje realizujące: Fondazione Parco Tecnologico Padano-Italy, UNIVERSITETET FOR MILJO OG BIOVITENSKA-Norway, THE UNIVERSITY OF EDINBURG-United Kingdom, UNIWERSYTET PRZYRODNICZY WE WROCLAWI-Poland, UNIVERSIDAD DE ZARAGOZ-Spain, ARISTOTELIO PANEPISTIMIO THESSALONIKI-Greece.
The Gene2Farm project will address the needs of the cattle industry, in particular of the SMEs and end users, for an accessible, adaptable and reliable system to apply the new genomic knowledge to underpin sustainability and profitability of European cattle farming. Gene2Farm will undertake a comprehensive programme of work from statistical theory development, through genome sequencing, to address new phenotyping approaches and the construction of tools, that will be validated in conjunction with SMEs and industry partners. Advanced statistical theory and applications will use the genomic and phenotypic information to optimise and customise genomic selection, breeding and population management and between breed predictions. The project will sequence key animals and exchange data with other international projects to create the most comprehensive bovine genome sequence database. Detailed analysis of these genome sequences will define genome structure, shared alleles, frequencies and historic haplotypes, within and between populations. This information will be used to optimise the informativeness of SNP panels and select SNPs to tag haplotypes, and hence ensure that genotype information can be used within and between breeds. The project will explore the opportunities for extended phenotypic collection, including the use of automated on farm systems and will develop standardisation protocols that, in consultation with ICAR, could be used by the industry for data collection and management. Developed tools will be tested and validated by demonstration in collaboration with dairy, dual purpose, beef and minority breed organisations. Finally a dissemination programme will ensure that training needs of the industry are served from an entry level training programme for farmers to advanced summer schools for the SMEs and expert user community.
Period of work: 2013
NADIR, The Network of Animal Disease Infectiology Research Facilities
Giulietta Minozzi, Joanna Szyda, Katarzyna Wojdak-Maksymiec
Genetic background of clinical mastitis in dairy cattle. The major goal of the project is to identify genomic features underlying differences in the level of mastitis resistance in Holstein-Friesian dairy cattle. In particular, using the animal material preselected based on the results of a previous study we aim to identify single nucleotide polymorphisms, copy number variations and indel polymorphisms which may influence the incidence of clinical mastitis.
Coordinated by prof. Dirk Hinrichs and prof. Georg Thaller
Major goals:
Factsheet
Results:
Preliminary results presented on the EAAP conference regarding Influence of pedigree on effective population size in European Red Dairy Cattle.
Economic values (in € per change in trait unit and cow-year), standard deviations and relative weights for the trait complex for the breeding goal traits for Polish dairy breeds: Polish Holstein (PH), Polish Red (PR).

Observed changes in revenues and costs due to a change of 1% increase in cow fertility expressed in € at herd level for Polish Red (PR) and Polish Holstein (PH).

Key investigators: Magdalena Frąszczak, Magda Mielczarek, Tomasz Suchocki, Joanna Szyda
Period: 2015-2018
The main goal of the project was to understand the genetic backgraound of clinical mastitis in dairy cattle. To achieve this goal we characterized the inter-individual variability resulting from the presence of SNP and CNV polymorphisms. As a result of the research, the following scientific articles were created:
Bioinformatics tools designed in this project have been additionally used to analyze the quality of the cattle reference genome (UMD 3.1), which is described in the article:
Moreover, these tools were used to describe breed specific SNPs in domestic cattle:
The major aims of this study is to use the whole genome DNA sequence of 32 individuals to identify mononucleotide polymorphisms as well as the Copy Number Variations (CNVs) which are present in the genome of domestic cattle. These polymorphisms will be then used to find genes or genomic regions responsible for the risk of clinical mastitis.
Whole genome DNA sequences are available for 32 cows representing the Polish Holstein-Friesian breed selected out of the data base of 991 cows comprising individuals with clinical mastitis cases diagnosed by a veterinarian and their healthy herdmates. The average sequencing coverage calculated across the 32 individuals is high and amounts to 14.03 varying between 5 and 17.
The experimental design comprises 16 paternal halfsib pairs comprising halfsisters matched by the number of parities, production level, and birth year, but differing in terms of their mastitis resistance expressed by the frequency of clinical mastitis diagnosed throughout their production life. In particular in each pair one of the halfsibs represents an animal without clinical mastitis occurrence throughout the whole production period (control group) and the other represents an animal with multiple clinical mastists cases (case group).
The alignment to the reference genome will be carried out based on a paired-end alignment, using the BWA-MEM software. Mononucleotide variant detection will be performed using the GATK and Samtools packages, further on the CNVnator program will be used for CNV detection, which uses local differences in read depth to identify copy number variation sites. Allele frequencies will be estimated using the Jackknife resampling algorithm. Moreover, the r2 statistics, which quantifies the amount of linkage disequilibrium between pairs of SNPs will be calculated using the PLINK package. SNP haplotypes will be reconstructed with GATK and Beagle software packages. The Odds Ratio and the Likelihood Ratio Tests will be used to assess differences in SNP allele frequency between case and control groups. Afterwards, the nominal P values will be corrected for multiple testing based using the estimated number of effective, independent tests. In addition, the False Discovery Rate will be calculated. For testing haplotype effects a logistic regression model will be compared with linear and quadratic discriminant models as well as with a random forest algorithm. Finally, a Variant Effect Predictor software together with custom written programs will be used for the functional annotation of the significant variants and regions.
Genetically, the significance of our project is related to the fact that, in contrast to commercial SNP panels, individual sequence data allow for the identification of rare genetic variants, which thanks to recent results from human genetic studies play a predominant role in the determination of genetic variation. To our knowledge no study involving rare variants has been conducted in dairy cattle. Also some novel statistical approaches need to be introduced: a novel method of multiple testing correction and a Jackknife resampling procedure, which has not been so far used in the context of whole genome DNA sequence analysis. Moreover, the understanding of genetic determination of clinical mastitis is of high importance for dairy cattle breeding, since udder infections in high performing cows are very common. They cause not only problems with animal welfare, but also considerable economical loss for the breeder.
The research was supported by the European Union Seventh Framework Programme through the NADIR (FP7-228394) project, by the Polish National Science Centre (NCN) grant 2014/13/B/NZ9/02016, and by The Leading National Research Centre (KNOW) programme for 2014-2018. Computations were carried out at the Poznan Supercomputing and Networking Centre
Key investigators: Joanna Szyda, Krzysztof Kotlarz
Period of work: 2020-2024
Funded: The National Science Centre (NCN)
Deep learning (DL) is a sub-field of machine learning methodology, which has recently and rapidly been gaining importance in many fields of science. Originally, it has been developed mainly for image recognition, but nowadays it has also been increasingly used in other fields, including genomics. According to the Editorial view of the Nature Genetics (January 2019) journal, deep learning algorithms are “to revolutionize genome analysis”. Their applications range from gene expression analysis, through modelling of gene expression regulation, to proteomics. However, in livestock genomics, analyses involving deep learning remain very sparse. Therefore, the goal of our project is to introduce the application of deep learning algorithms into this field.
In particular, our project is going to use deep learning for four different aspects of whole-genome DNA sequence analysis:
Next project’s results soon!









